DEFINITION GENERALITY
Amyloidosis was first described in the 19th Century.
Amyloidosis is now known to be a group of diseases in which one or more organ systems in the body accumulates protein deposits.
abnormal protein that may be deposited in any of your body's tissues or organs.
This abnormal protein comes from cells in the bone marrow.
disabling or life threatening.
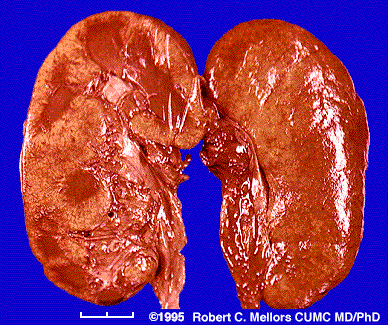
The disease known as amyloidosis (pronounced am-i-loy-do'-sis) results when enough amyloid protein builds up in one or more organs to cause the organ(s) to malfunction.
 The heart,
The heart, kidneys,
nervous system
spleen
adrenal
gastro-intestinal tract are most often affected.
the site of deposition depend on the type of amyloidosis primary or secondary.
Amyloidosis is a bone marrow disease.
bone marrow makes protective antibodies.
After they have served their function, these antibodies are broken down and recycled by the system.
In the amyloidosis, cells in the bone marrow produce antibodies that cannot be broken down.
These antibodies then begin to build up in the bloodstream.
Ultimately, they leave the bloodstream and can deposit in your tissues as amyloid.
amyloid is a hyalin substance that stains specifically with congo red, when examined with ME is found to be composed of fibers lacking periodicity [see specificity chapter]
amyloid is produced by the reticulum endothelial system and is deposited extracellularily.
recognition in 3 categories:
typical amyloidosis
slpeen
kidney
liver
adrenal
intestinal mucosa [rectal bipsy for diag]
atypical amyloidosis
any organ in addition to those mentioned [amyloidosis of the age affecting more the heart , 3% over 70 years old]
tumor forming amyloidosis
nodules of infiltrates by plasma cells may occurs [tongue]
tumor like deposits [pancreas islets and c cell thyroid tumors [calcitonin producing cells]
There are three major etiologic types, all very different from each other.
1. Primary Amyloidosis
familial mediterranean fever = typical amyloidosis
neuropathic amyloidosis = atypical amyloidosis
Primary amyloidosis is a plasma cell disorder and occasionally occurs with multiple myeloma.
This is the most common type of amyloidosis in the United States and is usually treated (conventionally) with chemotherapy.
Primary amyloid is not associated with any other diseases.
In primary amyloidosis, the organs most often involved include:
the heart, kidneys,
nervous system,
gastrointestinal tract.
Amyloid deposits in these organs cause:
shortness of breath,
fatigue,
edema (swelling of ankles and legs),
dizziness upon standing,
a feeling of fullness in the stomach (especially after eating),
diarrhea,
weight loss,
enlarged tongue,
numbness of the legs and arms,
and protein in the urine.
2. Secondary Amyloidosis
Secondary amyloid is caused by a chronic infection or inflammatory disease.
tuberculosis is present in 50% of the cases of amyloidosis
osteomyelitis 12%
chronic lung infection 10%
other chronic infections 12% = hyperimmunization
primary chronic polyarthritis 20% of cases show amyloidosis
Treatment of the underlying chronic infection or inflammatory disease can slow down or stop the progression of this type of amyloid.
In secondary amyloidosis, symptoms caused by the underlying chronic infection or inflammatory disease are complicated by the development of amyloid deposits in the kidney.
This may cause protein in the urine, edema, and fatigue
3. Hereditary Amyloidosis
Familial amyloid is the only type of amyloidosis that is inherited.
It is rare and is found in families of nearly every ethnic background.
In hereditary amyloidosis, the nervous system and gastrointestinal tract are often involved.
This can cause numbness and tingling in the arms and legs, dizziness upon standing, and diarrhea.
Families have their own pattern of organ involvement and symptoms.
The transmission is autosomal dominant, which means that if you have this type of amyloidosis each of your children has a 50% chance of inheriting the disease.
If your child does not inherit the gene, he/she cannot pass it on to future generations.
4. Other types of amyloidosis
include localized amyloid, b2 microglobulin amyloid, and Alzheimer's disease.
Localized types of amyloidosis are associated with hormones, proteins, aging, and specific areas of the body, and are not found with systemic involvment.
The type of amyloidosis which is due to the b2 microglobulin protein may affect people who have been on dialysis for a significant length of time.
In Alzheimer's disease, the amyloid protein in the brain is called the b-amyloid protein.
Amyloidosis can only be diagnosed by a positive biopsy; that is, an identification of the amyloid deposits in a piece of tissue.
Amyloidosis is rare, affecting about 8 persons per million annually. Its cause is not known.
It can affect anyone, but the majority of people who get amyloidosis are over the age of 40.
Primary amyloidosis is not contagious or inherited.
It is not known how many people have this disease.
About 10 percent of patients who have multiple myeloma (a form of bone marrow cancer), develop amyloidosis.
Although amyloid is an abnormal protein, diet and the amount of protein you eat play no role in the development of the disease.
Also, there is no recognized link between amyloidosis and stress or occupation.
ACQUIRED AMYLOIDOSIS
Protein
AL: Immunoglobulin light chains; k or l
Clinical
Most common in middle aged males
Associated disorders
 Paraproteinemia (M-protein)
Paraproteinemia (M-protein)  In 90% when serum & urine tested by immunofixation
In 90% when serum & urine tested by immunofixation Most common in nephrotic syndrome
Least common in polyneuropathy
Light chain in 1/3
Intact immunoglobulin in 2/3
l light chains: AL type
k light chains: Multiple myeloma; MGUS
Multiple myeloma : May present 10-81 months after diagnosis of AL
Polyneuropathy: Occurs in 20% of patients with light chain amyloid
Distal; Symmetric
Predominantly sensory
Pain & temperature loss most prominent
Distal weakness may develop later
Autonomic
Orthostatic hypotension
Hypoactive pupils
Hypohydrosis
Bladder dysfunction
Impotence
Carpal tunnel syndrome in 25%
Myopathy
Systemic features
Purpura: Periorbital
Submandibular swelling
Cardiac
Renal: Nephrotic syndrome
Muscle enlargement ± weakness
Gastrointestinal: Diarrhea
Anemia associated with multiple myeloma
Amyloidoma
Laboratory
CSF: Protein mildly elevated
Electrodiagnostic: Axonal neuropathy
Biopsy: Axonal loss; Amyloid
Prognosis
Gradual progression
Survival of 1 to 10 years depending on organ involvement
Shortest survival with cardiac (< 1 year) & renal dysfunction
 HEREDITARY PNS AMYLOIDOSIS
HEREDITARY PNS AMYLOIDOSISMutations in serum proteins that can form b-pleated sheets
Protein forming the amyloid is identified by immunocytochemistry
Liver transplantation may be effective therapy
ATTR: Transthyretin (Prealbumin)
 l Chromosome 18q11.2-q12.1; Dominant
l Chromosome 18q11.2-q12.1; Dominant ATTR gene
4 exons: Exon 1 codes for signal peptide
Mutations
General locations
Exon 1: Only 1 mutation (Val20Ile); Not in signal peptide
> 70 in other 3 exons
Specific mutations: Variable clinical presentation
Val30Met: Most common; Onset 25 to 65 years; Small fiber & autonomic D
Val28Met: 7th decade onset neuropathy with impotence; No FH
Database: HGMD
Types: Most are missense point mutations
ATTR Protein
127 amino acids
Present in plasma
Binds: Thyroxine (20%); Retinol binding protein
Clinical syndromes
Heterogeneous, with some relation to:
Point mutation & Ethnic background
Signs include:
Peripheral nerve
Polyneuropathy: Sensory, Autonomic, ± Motor
Carpal tunnel syndrome
Systemic: Cardiomyopathy, Vitreous opacities, Renal failure
Rare CNS
Glycine 18: Meningocerebrovascular syndrome sparing eyes
Glycine 30: Oculoleptomeningeal syndrome
Ile107Val: Autonomic; Carpal tunnel; CNS; Multisystem (Cardiomyopathy; Lung; Joint; Angiopathy)
General signs: Hearing loss; Migraine; Dementia; Cerebellar; Seizures; Stroke; Myelopathy
Onset 17-78 years
Earlier: Portuguese; Japanese endemic Val30Met foci
Later: Swedish; Sporadic Japanese Val30Met patients
? Anticipation: Not explained by triplet repeats
Homozygotes & heterzygotes with same clinical manifestations
Penetrance: Variable: 30% to 95%
FAP I
Presentation: Generalized Autonomic & Sensory Polyneuropathy
Onset: Legs
Painful dysesthesias
Most common point mutation: Val30Met
Onset: 20 to 80 years
Later: Sporadic Japanese cases; Swedish endemic foci
Earlier: Japanese & Portugese endemic foci
Penetrance
High: Endemic Japanese foci
Low: Scattered, sporadic cases; Swedish endemic foci
Male > Female: Especially in late onset sporadic cases
Weakness: Distal; Legs > Arms
Sensory loss
Early onset: Pain & Temperature loss most prominent
Late onset: Distal; Symmetric; All modalities; Paresthesias
Tendon reflexes: Reduced or Absent
Autonomic failure
Severe in early onset patients
Mild in late onset patients
Progression: Death in 7 to 10 years
CSF protein: Moderately é in 20%
Sural nerve biopsy
Axonal loss: Myelinated axons especially in older onset patients
Amyloid: Most prominent in sympathetic ganglia, dorsal root ganglia & proximal nerve trunks
Treatment: Liver transplantation
Also > 23 other mutation
"Portuguese, Swedish & Japanese" types
FAP II
Onset:
Upper extremities, esp carpal tunnel syndrome
5th decade
Most common mutation: Ser77Tyr
Onset
51 to 67 years
Carpal tunnel syndrome: May be only manifestation for 1 to 2 decades
Generalized polyneuropathy
Pain & Paresthesias
Weakness: Symmetric; May become severe
Restrictive cardiomyopathy: Arrhythmia; Cardiac insufficiency
Autonomic: Intestinal malabsorption; Hypotnesion
No renal or ocular involvement
Progression
Variable: Some stable; Others rapidly progressive
Survival high
Severe disability may develop
Treatment: Liver transplantation for disabled < 65 years
> 6 other point mutations
"Indiana & Maryland " types
Other point mutations present with Cardiomyopathy or Renal disease
Cardiomyopathy: Val122Ile mutation
Common in black population
Late onset: > 60 years
AApoA1: Apolipoprotein A-1 (FAP III; "Iowa" type)
l Chromosome 11q23.3; Dominant
Gene
Neuropathic amyloid: Point mutation Gly26Arg
Other amyloid mutations: Trp50Arg; Insertions & deletions
Protein
Transports cholesterol from tissues to liver
Major plasma & chylomicron protein
Clinical features (Iowa type): Similar to FAP I
Polyneuropathy
Nephropathy
Gastric Ulcer
Other non-neuropathic amyloid syndromes
Hepatic failure
Cardiac ± Cutaneous: Leu90Pro; Arg173Pro; Leu174Ser
AGel: Gelsolin (FAP IV; "Finnish" (also Japanese) type )
l Point mutations amino acid 187; Chromosome 9q32-q34; Dominant
Protein
Binds to:
Actin: barbed end of filaments; Prevents monomer exchange
Fibronectin
Intracellular: Muscle; Phagocytes; Fibroblasts; Platelets
Clinical features
Onset: 4th decade
Cranial Neuropathies
Facial paresis (Upper)
Also: V; XII; VIII
Corneal lattice dystrophy: Also see Lattice corneal dystrophy IIIA
Sensory neuropathy (Vibration loss) ± autonomic
Skin: Laxity
No cardiac involvement
Homozygotes: Rapidly progressive disease & Renal failure
What are the symptoms of amyloidosis in general?
Symptoms of amyloidosis depend on the organs it affects. The wide range of symptoms often makes amyloidosis difficult to diagnose. You may have no symptoms. Symptoms can include:
Swelling of ankles and legs
Weakness
Weight loss
Shortness of breath
Numbness or tingling in the hands or feet
Diarrhea
Severe fatigue
Enlarged tongue
Feeling of fullness after eating smaller amounts of food than usual
Dizziness upon standing
How is amyloidosis diagnosed?
Physical examination is necessary to find out if your organs are functioning properly.
Blood, urine and bone marrow tests may also be done.
A small tissue sample (biopsy) may be taken from your rectum, abdominal fat or bone marrow to determine if you have amyloidosis.
Occasionally, samples are taken from the liver, nerve, heart or kidney.
Blood or urine tests can detect the protein, but only bone marrow tests or other small samples of tissue can positively establish the diagnosis of amyloidosis